LINKZOL ®官网|人工智能|高性能计算领域服务器设备及OEM定制品牌厂商
LINKZOL ®官网|人工智能|高性能计算领域服务器设备及OEM定制品牌厂商 |
 :400-630-7530 :400-630-7530
|
 :sales@linkzol.com :sales@linkzol.com |
- 产品信息查询 - |
|
LINKZOL ®官网|人工智能|高性能计算领域服务器设备及OEM定制品牌厂商 |
:400-630-7530
|
:sales@linkzol.com |
- 产品信息查询 - |
|
|
解决方案
|
|
|
分子动力学模拟也能用GPU加速?NVIDIA Tesla告诉你答案AMBER(Assist Model Building with Energy Minimization)是一款著名的分子动力学模拟软件包。从 V11 版本开始,AMBER 开始可以使用 NVIDIA 的 GPU 来进行显性溶剂 PME 和隐性溶剂 GB 仿真。 本测试针对 Cellulose(纤维素)进行,确认AMBER在 GPU 上的加速效果。 默认情况下,AMBER 不会自动去调用 GPU,于是我们手动编译了支持 cuda 的版本, pmemd.cuda.MPI(多 GPU 版)与 pmemd.cuda(单 GPU 版) 。 在本次测试中所使用的分子是纤维素(Cellulose),它的原子数量为:408,609 atoms。 测试步骤
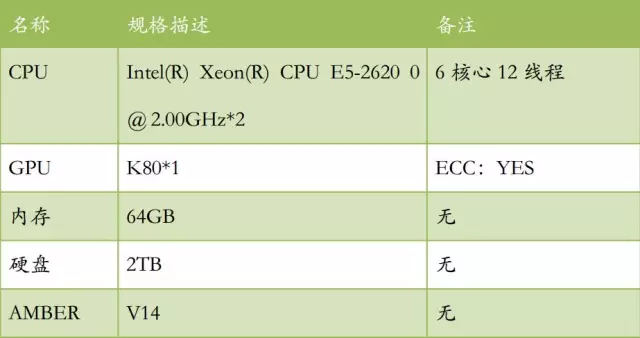
测试环境
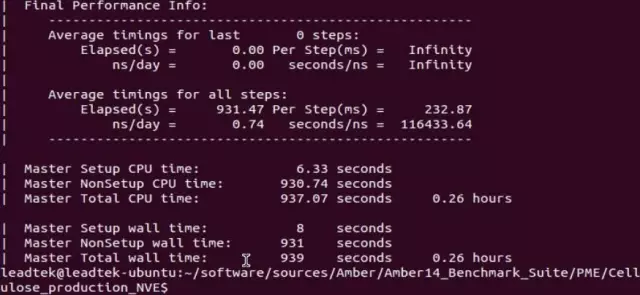
在 CPU 下的测试
本次测试中所使用的分子是纤维素(Cellulose),它的原子数量为:408,609 atoms。 使用 CPU 编译的版本,在 CPU 运行的测试结果如下图所示。
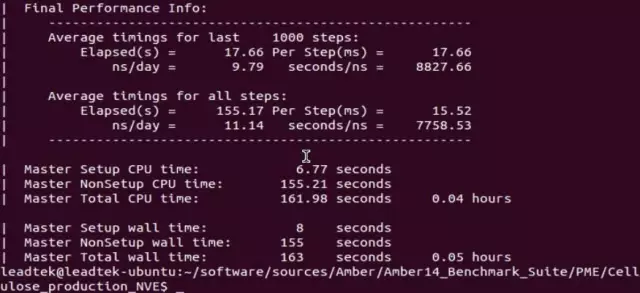
在单 GPU 下的测试
在双 GPU 下的测试
在本次测试中使用双 GPU 来进行纤维素的分子模拟,需要使用多 GPU 支持的并行版本,使用 pmemd.cuda.MPI 命令,每个进程推荐使用一个 GPU。其最后的测试结果如下图所示。
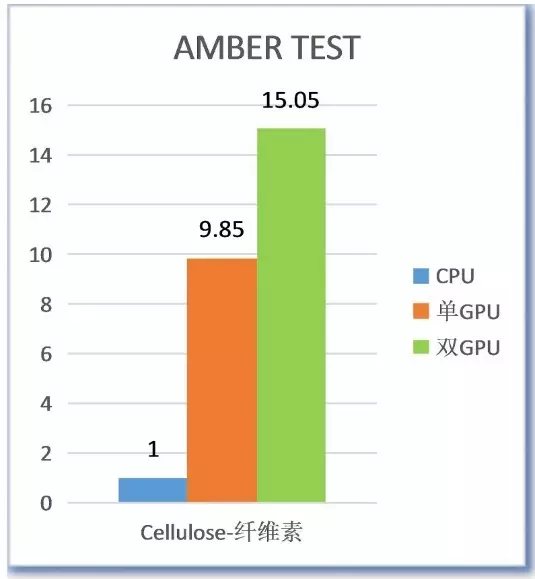
测试结果
测试结果证明NVIDIA Tesla GPU加速器对AMBER有极大幅度的提升。 推荐配置 |
|